|
11月8日,清华大学化工系生物育种技术与装备团队报道了一种高精度的对蛋白表达特征进行大规模并行表征的方法,该方法可应用于对混合文库中各个基因型对应的蛋白表达均值和异质性的高通量表征。
细胞内基因表达是将遗传信息从基因传递到RNA,然后再传递到蛋白质的过程,很大程度上决定了细胞的性状。然而,基因表达常常表现出随机性,因为它涉及到多种低拷贝分子的随机参与,包括聚合酶、核酸、转录因子、核糖体等。此外,由于生物调控网络的高度复杂性和非线性,基因表达可能会表现出混沌特征,即对环境和代谢物的微扰十分敏感。因此,即使在相同的环境条件下,基因型完全相同的细胞之间也存在表型差异,这种现象被称作表型异质性。与此相对应的是,当在同一环境中培养的单克隆细胞群体中,针对每个个体的蛋白质表达量在某一时间点进行统计时,会观察到一种分布。这个分布的平均值表示表达的强度,而变异系数则代表表达的噪声水平。这两个特征与种群的表型密切相关,例如代谢产物的生成量、药物抗性和持留性等。然而,目前对这两个特征的同时定量研究往往依赖于使用流式细胞仪或者荧光显微镜对单克隆种群进行一一测试,十分耗时耗力。为解决这个问题,研究团队针对流式分选-测序实验框架和数据产生过程提出了深度学习辅助的流式分选-测序方法(dSort-Seq),可实现对基因表达特征的高通量并行表征。
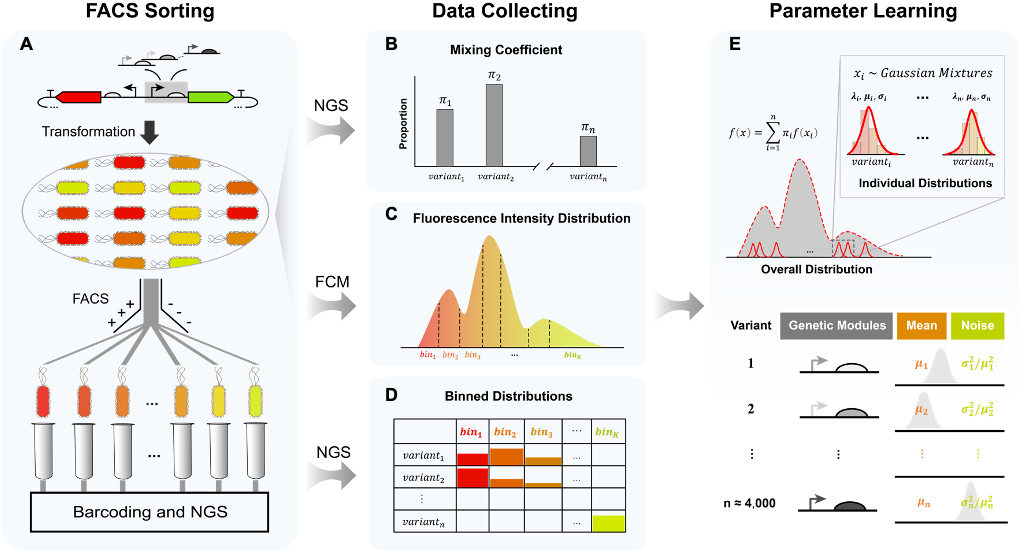
流式分选-测序方法及数据处理流程
在该方法中,研究团队提出使用双成分对数混合高斯(LGMM)来表示单克隆种群基因表达分布。相较于传统的伽马分布和对数正态分布,LGMM体现出更高的精确度和鲁棒性。随后,研究团队根据流式分选-测序实验的数据生成过程,构建了贝叶斯神经网络进行参数学习,用于表达特征的计算。
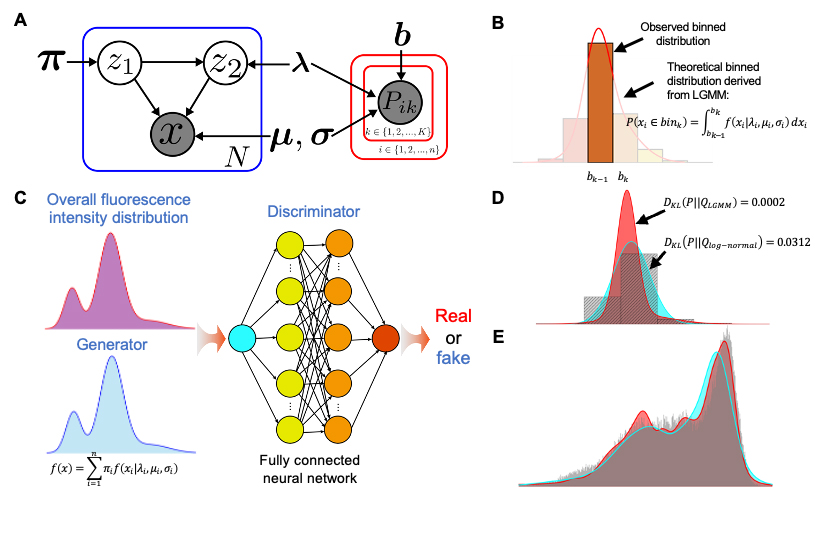
模型的图表示和参数学习
研究团队首先使用课题组先前报道的流式分选-测序数据验证了dSort-Seq的可靠性,并将该方法应用于计算丙二酰辅酶A生物传感器组合文库在不同效应物浓度下的响应大小,得到的结果与单独流式测试结果一致。证明该方法可以应用于生物传感器剂量-响应曲线的高通量表征。
此外,研究团队对大肠杆菌中转录和翻译对表达噪声的贡献进行了深入探究。为此,他们构建了大肠杆菌内源启动子文库(库容量3804)和启动子RBS组合文库(300个启动子和13个RBS,库容量3900),并使用dSort-Seq对两个文库的表达特征进行定量研究。研究发现转录与表达噪声强度(Fano因子)存在正相关关系,且转录和翻译对噪声的贡献大小基本相同,这与传统的翻译爆发机制相悖。进一步的,研究团队发现呈现出高噪声表型的启动子往往具有高的T碱基含量,而这一现象可能与重叠的RpoD识别位点有关。最后,研究团队证明重叠的RpoD识别位点会引发高的表达噪声,预示了一种新的噪声调控方案。
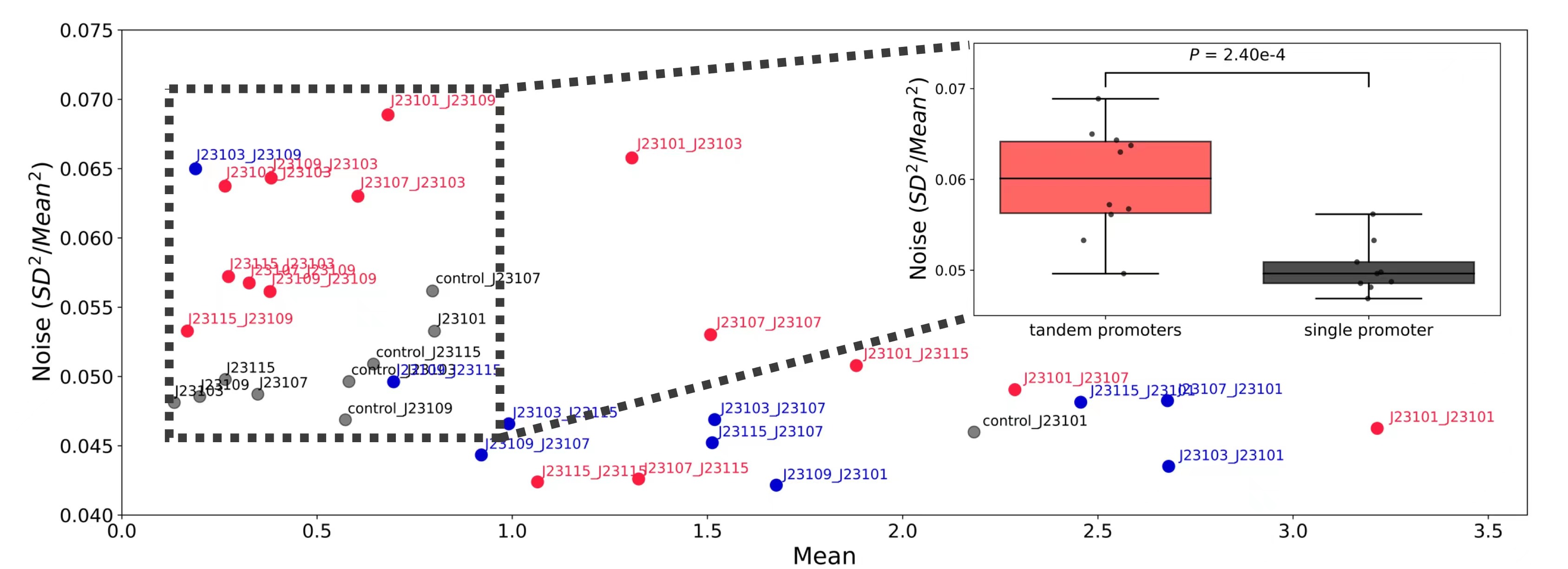
重叠的RpoD识别位点引发高表达噪声
该工作以研究论文的形式于11月8日在《科学进展》(Science Advances)上发表。化工系张翀教授和2018级博士冯汇宝(现为加州理工学院博士后)为该文章的共同通讯作者,化工系冯汇宝和2019级硕士李帆(现为苏黎世联邦理工学院博士)为该文章的共同第一作者,清华大学深圳国际研究生院生物医药与健康工程研究院常务副院长邢新会教授、西湖大学合成生物学和生物工程讲席教授曾安平和上海科技大学王天民研究员为本文共同作者。该成果得到了国家重点研发计划、国家自然科学基金、佛山-清华创新专项基金和湖北大学生物催化与酶工程国家重点实验室开放课题基金的资助。
论文链接:https://www.science.org/doi/10.1126/sciadv.adg5296
清华大学化工系

|